{kind=link}
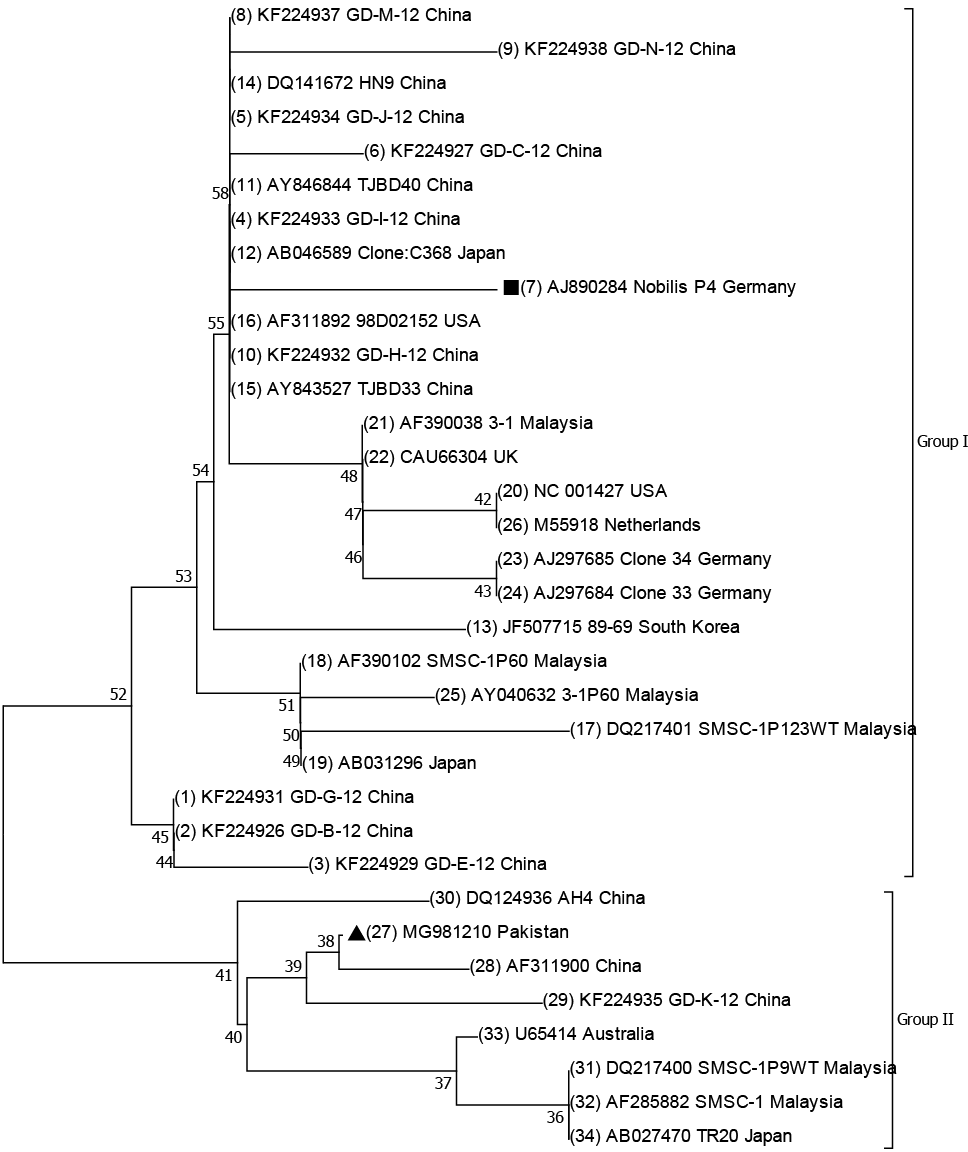
The phylogenetic tree was inferred by the Neighbor Joining method as described by Saitou and Nei, 1987. The optimal length of the tree with sum of branch length = 0.11748525 is shown. The tree is sketch to scale having branch lengths in the same units which are used in evolutionary distances to infer the phylogenetic tree. Evolutionary distances were calculated by using the Poisson correction method as mentioned by Zuckerkandl and Pauling (1965) and are in the units of the number of amino acids swipe per site. The analysis is made from thirty-four amino acids sequences. All sites having gaps and missing data were eliminated. There were total 222 positions in final dataset. Evolutionary assessment was carried out in MEGA7 (Kumar et al., 2016).